
Vascularite
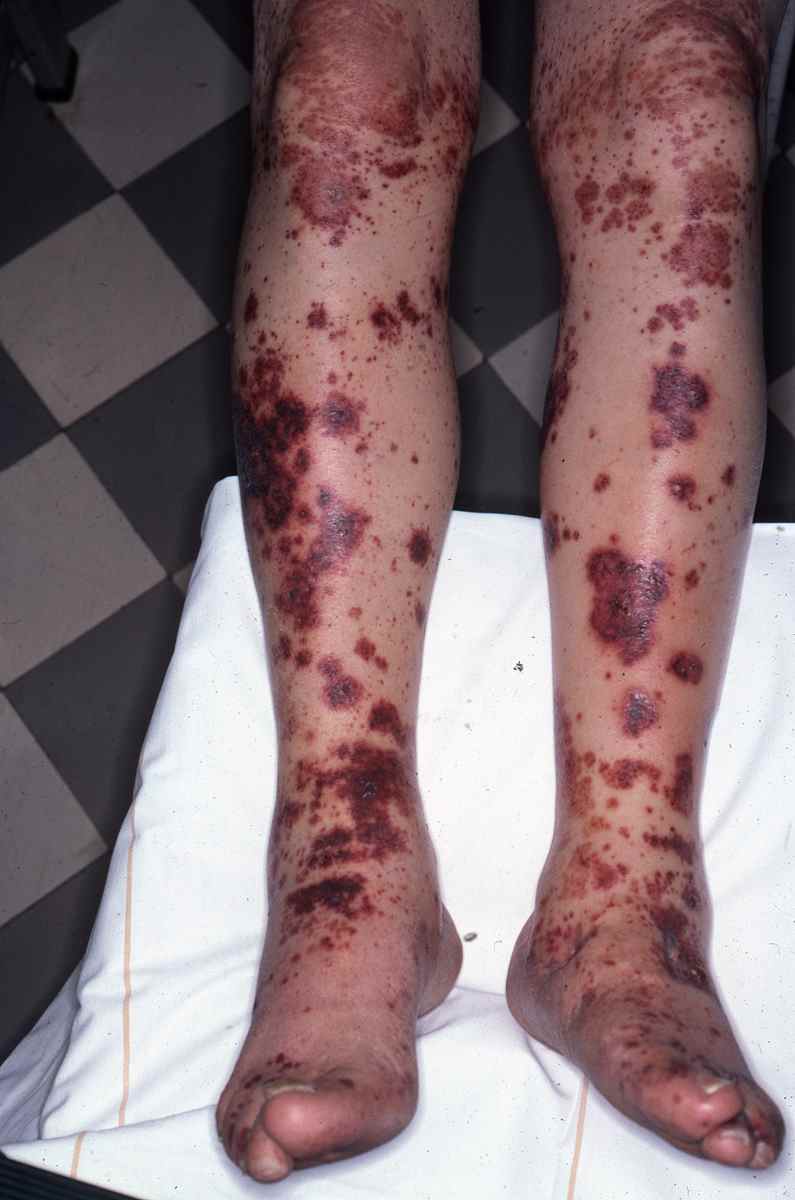
La vascularite, encore appelée vasculite ou angéite, est une inflammation des parois des vaisseaux aboutissant à des dégats de la paroi vasculaire.
Elle peut concerner les gros vaisseaux (aorte et ses branches), jusqu’aux capillaires, veinules et artérioles (vascularite des petits vaisseaux). Entre deux se situe la vascularite des moyens vaisseaux. On la découpe donc en trois groupes en fonction du diamètre des vaisseaux concernés.
Classification des vascularites
A/ Vascularite des gros vaisseaux (aorte et ses branches)
B/ Vascularite des moyens vaisseaux
B.1/ Péri Artérite noueuse ou PAN
C/ Vascularites des petits vaisseaux (capillaires, artérioles, veinules)
C.1/ Maladie du Sang et le la moelle
Leucémie, lymphome, myélome..
C.2/ Infection
- bactérienne : streptocoque, cocci à Gram négatif (gonocoque et méningocoque)
- virale : hépatites, MNI,VIH…
- parasitaire : paludisme…
- fongique : Candida albicans…
C.3/ Néoplasie
C.4/ Iatrogène
- médicament : antibiotiques (pénicillines, sulfamides, quininolones… ), allopurinol, diuretiques thiazidiques, rétinoïdes, cytokines…
- vaccin…
C.5/ Idiopathique dans 50% des cas…
C.6/ Auto immun et immunologique
- Cryoglobulinémies de type II (mixtes monoclonales) et III (mixtes polyclonales), associées à une maladie auto-immune, à une infection (surtout hépatite C) ou à une hémopathie
- Hypocomplémentémie (vascularite urticarienne de Mac Duffie)
- Hyperglobulinémie (purpura hyperglobulinémique de Waldenström)
- Connectivites : lupus, syndrome de Gougerot-Sjögren, polyarthrite rhumatoïde…
- Déficit en α1-antitrypsine
- Vascularites associées aux ANCA (anticorps anti cytoplasme des polynucléaires neutrophiles) :
A/ Vascularite des gros vaisseaux (aorte et ses branches)
A.1/ Maladie de Horton
A.1.1/ Symptomes
La maladie de Horton est une vascularite d’origine mal connue atteignant les personnes de plus de 60 ans. La vascularite concerne le réseau carotidien externe : les signes sont donc ischémiques au visage et sur le cuir chevelu (augmentation de volume et douleur des artères temporales, claudication mandibulaire, nécroses de la langue et des lèvres ou du cuir chevelu, maux de tête, douleurs et une hypersensibilité du cuir chevelu) .
L’atteinte ophtalmologique (baisse d’acuité visuelle, paralysie oculo-motrice, ptosis, anomalie pupillaire) peut être grave et mener à la cécité par névrite optique ischémique généralement irréversible.
L’ensemble survient souvent dans un contexte d’altération de l’état général, de fièvre, d’anomalies vasculaires (claudication d’un membre, anévrysmes artériels inflammatoires aortiques, accidents vasculaires cérébraux, ischémie myocardique ou digestive, syndrome de Raynaud), rhumatologiques (pseudo-polyarthrite rhizomélique) ou respiratoires (toux sèche inexpliquée).
A.1.2/ examens
Syndrome inflammatoire majeur (vitesse de sédimentation supérieure à 80 mm à la 1ère heure).
Echographie couleur-Doppler artériel : anomalies de flux artériel associées à un halo inflammatoire péri-artériel.
Biopsie pour examen anatomopathologique au microscope d’un fragment artériel, le plus souvent l’artère temporale superficielle d’une taille d’au moins 3 cm.
A.1.3/ Traitement
Corticothérapie par voie orale au long cours, d’où des risques chez les personnes agées
A.2/ Maladie de Takayasu
La maladie de Takayasu ou « maladie des femmes sans pouls » est une vascularite d’origine mal connue, touchant tout l’arc aortique et ses branches (artères coronaires, carotides, sous-clavières, vertébrales, rénales, iliaques) ainsi que des artères pulmonaires d’où son nom de « syndrome de l’arc aortique« .
Cette maladie rare touche surtout la femme jeune entre 20 et 35 ans. On la rencontre plus fréquemment en Asie.
A.2.1/ Symptomes
On distingue la période aiguë, dite pré-occlusive et la période occlusive.
A.2.1.1/ Période pré-occlusive
Cette période est caractérisée par l’apparition de signes généraux peu spécifiques (fièvre, douleurs articulaires, altération de l’état général, amaigrissement… ).
Parfois on constate des douleurs de l’œil, des troubles de la vision, un érythème noueux
Puis apparaissent des douleurs cervicales le long du trajet de l’artère carotide (carotinodynies) plus évocatrices de la maladie, des douleurs des membres supérieurs, une faiblesse ou une fatigabilité plus prononcée d’un côté à l’autre.
A.2.1.2/ Période occlusive
A ce stade les artères deviennent rétrécies (sténoses) ou bien occluses. On peut s’en rendre compte fortuitement lors de la prise de la pression artérielle différente entre les 2 bras ou une absence de pouls par endroits.
A.2.2/ Examens complémentaires
L’échographie-couleur Doppler artérielle, l’angio-scanner, ou encore l’angio-IRM, le TEP-scanner (tomographie par émission de positons) et l’artériographie montrent des sténoses en « flamme de bougie » ou en « queue de radis« , des occlusions artérielles ou des anévrysmes, des inflammations de la paroi de l’aorte (aortite).
2.3/ Traitement médical
Il repose sur la corticothérapie durant un à deux ans parfois associé à un traitement immunosuppresseur (méthotrexate, azathioprine, mycophénolate mofétil).
En cas de résistance au traitement, des biothérapies peuvent être tentées : anti-TNF (Tumor Necrosis Factor) (infliximab) ou anti-récepteurs de l’interleukine 6 (tocilizumab)
B/ Vascularite des moyens vaisseaux
B.1/ Péri Artérite noueuse ou PAN
La périartérite noueuse (PAN) est une vascularite nécrosante très rare pouvant concerner de nombreux organes, dont la cause est mal connue (certaines formes seraient liées au virus de l’hépatite B).
B.1.1/ Symptomes
B.1.1.1/ Signes généraux
Les patients ont souvent une altération de l’état général avec amaigrissement, fièvre…
B.1.1.2/ Douleurs musculaires
Les douleurs musculaires sont présentes dans la moitié des cas. Elles sont intenses, diffuses, spontanées ou déclenchées par la pression, pouvant clouer le patient au lit du fait l’intensité des douleurs et de la fonte musculaire…
B.1.1.3/ Douleurs articulaires
Les douleurs prédominent sur les grosses articulations périphériques : genoux, chevilles, coudes et poignets.
B.1.1.4/ Manifestations neurologiques
On constate souvent une multinévrite (inflammation à la fois de plusieurs nerfs) tels que le sciatique, le poplité externe ou interne, les nerf radial, cubital ou médian et elles sont souvent associées à un œdème segmentaire distal puis une atrophie des muscles innervés par le nerf concerné.
La vascularite peut aussi concerner le cerveau plus rarement, pouvant donner une épilepsie, une hémiplégie, des accidents vasculaires cérébraux, ischémiques ou hémorragiques.
B.1.1.5/ Manifestations cutanées
Le signe évocateur de vascularite est le purpura (taches violacées qui ne s’effaçent pas lorsqu’on appuie dessus) infiltré, surtout aux membres inférieurs ou un livedo, formant des sortes de mailles (livedo reticularis) ou marbrures (livedo racemosa) violacées sur les jambes. On peut aussi constater un phénomène de Raynaud (quelques doigts blanchissent au froid), voire des gangrènes de doigt ou d’orteil.
B.1.1.6/ Manifestations uro génitales
L’orchite (inflammation d’un testicule) est une des manifestations les plus typiques de la PAN, due à une vascularite de l’artère testiculaire pouvant aboutir à une nécrose testiculaire.
B.1.1.7/ Manifestations cardiovasculaires
La PAN provoque une insuffisance cardiaque et une hypertension artérielle pouvant être graves.
B.1.1.8/ Manifestations digestives
1/3 des patients atteints de PAN ont des douleurs abdominales dues à une vascularite du tube digestif pouvant évoluer en hémorragie digestive voire en une perforation de l’intestin grêle.
Critères de diagnostic de la périartérite noueuse selon l’American College of Rheumatology
- Amaigrissement > 4kg
- Livedo reticularis
- Douleur ou sensibilité testiculaire
- Myalgies diffuses, faiblesse musculaire ou sensibilité des membres inférieurs
- Mono- ou polyneuropathie
- Pression diastolique > 90 mmHg
- Insuffisance rénale (urée > 14,3 mmol/l ou créatininémie > 132 μmol/l)
- Présence de l’antigène HBs ou de l’anticorps anti-HBs dans le sérum
- Anomalie artériographique (anévrysme et/ou occlusion des artères viscérales)
- Biopsie d’une artère de petit ou moyen calibre montrant la présence de polynucléaires dans la paroi artérielle
Chez un sujet atteint de vascularite, la présence de 3 des 10 critères diagnostique une périartérite noueuse dans 85% des cas.
B.1.2/ Examens complémentaires
Un syndrome inflammatoire est présent chez la majorité des patients atteints de PAN (augmentation de la vitesse de sédimentation à plus de 60 mm à la première heure, de la C Réactive Protéine… ), ainsi qu’une hyper éosinophilie majeure (augmentation des globules blancs polynucléaires éosinophiles).
L’infection par l’hépatite B se traduit par la présence de l’antigène HBs chez environ ¼ à 1/3 des patients
L’angiographie révèle des microanévrysmes et des sténoses (diminution de calibre ou aspect effilé) de vaisseaux de moyen calibre.
B.1.3/ Traitement
Le traitement de la PAN commence par la corticothérapie, parfois associée à des immunosuppresseurs (cyclophosphamide notamment)
Les biothérapies commencent à prendre place dans la prise en charge de la PAN, notamment le rituximab (anti-CD20).
B.2/ Maladie de Buerger
La maladie de Buerger ou thromboangéite oblitérante est une vascularite touchant des parties d’ artères de petit et moyen calibre et de veines des membre inférieurs et supérieurs, provoquant une thrombose et une recanalisation des vaisseaux atteints.
Cette maladie est plus fréquente en Asie et chez les juifs ashkénazes.
Elle survient chez un patient d’age inférieur à 45 ans, généralement fumeur (le tabac est un facteur déclencheur et aggravant de la maladie), présentant des manifestations d’artérite tôt dans la vie (ischémie des doigts ou orteils, claudication intermittente, ulcères artériels ischémiques ou gangrènes des jambes… )
L’artériographie révèle des atteintes des artères distales.
Le traitement passe par l’arrêt total du tabac, des vasodilatateurs et des antiagrégants plaquettaires tels que l’aspirine voire de la chirurgie de revascularisation.
B.3/ Maladie de Kawasaki
La maladie de Kawasaki ou « syndrome adéno-cutanéo-muqueux » est une vascularite touchant électivement les artères coronaires provoquant des anévrysmes coronariens qui peuvent être source de mortalité ou de lourdes séquelles cardiaques
Cette vascularite se rencontre chez l’enfant entre 6 mois et 5 ans avec un pic à l’âge de 18 mois.
B.3.1/ Symptomes
La vascularite se déroule en trois phases sur plusieurs semaines
1. Phase aiguë (durant 7 à 14 jours)
fièvre avec une éruption cutanée et aspect de « lèvres gonflées cerise« , « langue fraise« , « yeux injectés » par une conjonctivite bilatérale, « enfant inconsolable« , œdème et rougeur des mains et pieds.
Idéalement le traitement doit être mis en place à ce stade pour limiter le risque de séquelles cardiaques
2. Phase subaiguë (14 à 28 jours)
aboutissant à une desquamation de la pulpe des doigts et des orteils débutant autour des ongles.
Les anévrysmes coronariens se forment à cette période surtout
3. Phase de convalescence
en principe sans symptomes, mais durant laquelle des complications cardiaques subites peuvent survenir en raison de la formation d’anévrysmes coronariens dans la phase précédente.
Le diagnostic repose sur six critères principaux répertoriés par le Japan Kawasaki Disease Research Commitee :
- fièvre de plus de 5 jours ;
- conjonctivite bilatérale ;
- modification des extrémités : rougeurs des paumes et des plantes, œdème induré des mains et des pieds et desquamation péri-unguéale ;
- modifications oro-pharyngées : rougeur, fissures et gonflement des lèvres et « langue fraise » ;
- eruption (« rash ») polymorphe ;
- ganglion du cou
Les autres signes sont
l’érythème fessier, rouge vif avec une collerette desquamative,
signes cardio-vasculaires (souffle au coeur, galop cardiaque, anomalies à l’Electro CardioGramme, péricardite, myocardite… ),
signes digestifs (diarrhée, vomissements, douleurs abdominales… ),
neurologiques (méningite aseptique, convulsions, paralysie),
urinaires (pus stérile dans les urines, urétrite),
polyarthrite…
B.3.2/ Examens complémentaires
Inflammation importante avec une Vitesse de Sédimentation supérieure à 100mm à la première heure et une protéine C-réactive très élevée, une augmentation franche des globules blancs polynucléaires supérieure à 20 000 éléments/mm3, et une augmentation des plaquettes.
B.3.3/ Traitement
Le traitement repose sur les immunoglobulines injectées en Intra-Veineuse (Ig IV) le plus tôt possible pour limiter le risque d’anévrysme coronarien. En l’absence d’efficacité des IgIV, on peut utiliser de la cortisone en injection intraveineuse ou de l’aspirine.
C/ Vascularites des petits vaisseaux (capillaires, artérioles, veinules)
C.1/ Symptomes
C.1.1/ Signes cutanés
L’aspect caractéristique des vascularites est le purpura infiltré, palpable, prédominant aux jambes, aggravé par la position debout, pouvant prendre plusieurs formes (pétéchial et ecchymotique, nécrotique, pustuleux… )

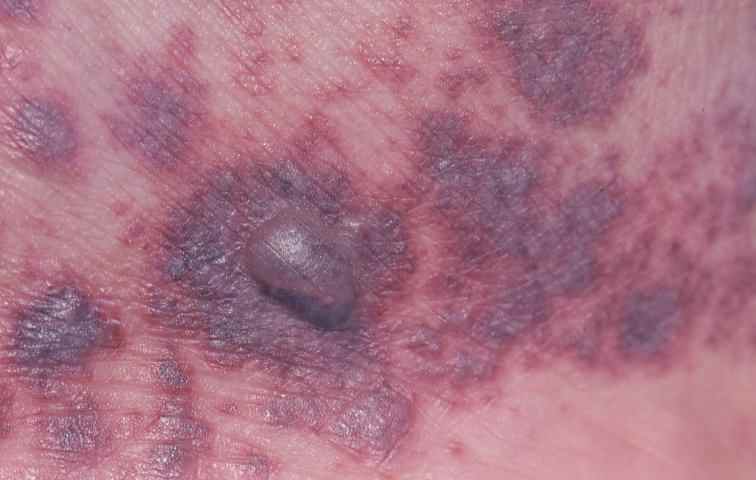
Le purpura peut être associé à d’autres lésions sur la peau (papules, nodules, lésions nécrotiques, bulles hémorragiques, urticaire fixe qui ne démange pas ou livedo).
C.1.2/ Manifestations systémiques, signes de gravité
La présence de manifestations en dehors de la peau constitue un facteur de gravité, montrant la présence d’une atteinte vasculaire au niveau des organes :
douleurs articulaires,
douleurs abdominales, selles noires, trouble du transit,
neuropathie périphérique
œdèmes des membres inférieurs,
Hyper Tension Artérielle,
difficultés à respirer, asthme, crachats de sang…
C.2/ Examens complémentaires
Les examens ont pour but de rechercher une cause et des signes de gravité
Le médecin prescrit systématiquement un bilan de purpura infiltré avec doute de vascularite :
– NFS, VS, TP, TCA, fibrinémie
– bilan hépatique : transaminases, γ-GT, bilirubine, phosphatases alcalines
– fonction rénale : urée et créatinine, protéinurie des 24 heures et hématurie (ECBU)
– bilan immunologique : complément, facteurs antinucléaires, latex, Waaler-Rose, électrophorèse des protéines, immuno-électrophorèse, cryoglobuline, ANCA, anticorps antiphospholipides
– sérologies : ASLO, hépatites B et C ± VIH
– recherche de sang dans les selles
En cas de point d’appel clinique ou selon le contexte, il peut prescrire aussi :
– bilan radiologique : radiographie pulmonaire en cas de dyspnée…
– bilan infectieux : hémocultures (même sans fièvre en cas de valvulopathie), prélèvement de gorge, prélèvement urétral…
– biopsie rénale : en cas de protéinurie supérieure à 1 g/24 h à plusieurs reprises ou en cas d’altération de la fonction rénale
– test de Schirmer et test au sucre en cas de syndrome sec
C.3/ Classement des causes de vascularites des petits vaisseaux
C.3.1/ Maladie du Sang et le la moelle
Leucémie, lymphome, myélome..
C.3.2/ Infection
- bactérienne : streptocoque, cocci à Gram négatif (gonocoque et méningocoque)
- virale : hépatites, MNI,VIH…
- parasitaire : paludisme…
- fongique : Candida albicans…
C.3.3/ Néoplasie
C.3.4/ Iatrogène
- médicament : antibiotiques (pénicillines, sulfamides, quininolones… ), allopurinol, diuretiques thiazidiques, rétinoïdes, cytokines…
- vaccin…
C.3.5/ Idiopathique dans 50% des cas…
C.3.6/ Auto immun et immunologique
- Cryoglobulinémies de type II (mixtes monoclonales) et III (mixtes polyclonales), associées à une maladie auto-immune, à une infection (surtout hépatite C) ou à une hémopathie
- Hypocomplémentémie (vascularite urticarienne de Mac Duffie)
- Hyperglobulinémie (purpura hyperglobulinémique de Waldenström)
- Connectivites : lupus, syndrome de Gougerot-Sjögren, polyarthrite rhumatoïde…
- Déficit en α1-antitrypsine
- Vascularites associées aux ANCA (anticorps anti cytoplasme des polynucléaires neutrophiles) :
La micropolyangéite (MPA) est une vascularite nécrosante systémique dont les signes cliniques sont très proches de celles de la PAN.
La MPA est associée aux ANCA de type anti-méloperoxydase (anti-MPO) et elle provoque classiquement une glomérulonéphrite rapidement progressive et une atteinte pulmonaire qui est absente dans la PAN.
Le traitement de la MPA comme pour la PAN commence par la corticothérapie, parfois associée à des immunosuppresseurs (cyclophosphamide notamment)
La granulomatose de Wegener est une vascularite dont le début est généralement marqué par des symptômes ORL ou respiratoires résistant aux traitements antibiotiques
Classiquement, une atteinte ORL grave (pansinusite destructrice), pulmonaire (nodules parenchymateux) et rénale (glomérulonéphrite nécrosante pauci-immune à croissants) est la triade de la granulomatose de Wegener.
L’atteinte cutanée et muqueuse concerne environ la moitié des patients : purpura infiltré, papules, nodules sous-cutanés, ulcérations cutanées, pustules, vésicules, gingivite hyperplasique.
Critères de classification de l’American College of Rheumatology pour la granulomatose de Wegener.
- Inflammation orale ou nasale
- Radiographie thoracique anormale (nodules pleins ou excavés, infiltrats fixes)
- Hématurie microscopique
- Granulome vasculaire, périvasculaire ou extravasculaire
La présence d’au moins deux critères est requise + les c-ANCA et/ou p-ANCA.
Le traitement de la granulomatose de Wegener, souvent considérée comme une urgence médicale, doit s’effectuer en milieu hospitalier par cortisone (prednisone à la dose initiale de 1 mg/kg/j) et d’un agent cytotoxique tel le cyclophosphamide par voie orale ou de méthotrexate administré de manière hebdomadaire (15 à 25 mg/semaine).
Cette vascularite granulomateuse est caractérisée par un asthme grave corticodépendant avec hyperéosinophilie dans plus de 90% des cas, précédant en moyenne de 8 ans les premiers signes de la vascularite et persistant après sa guérison.
Critères de classification de l’American College of Rheumatology (1990).
- asthme
- éosinophilie sanguine supérieure à 10 %
- mono- ou polyneuropathie
- infiltrats pulmonaires labiles
- douleur ou opacité sinusienne
- présence d’éosinophiles extravasculaires sur la biopsie
La présence de 4 des 6 critères permet un diagnostic de maladie de Churg et Strauss approximativement à 90%
Le traitement du syndrome de Churg et Strauss est identique à celui de la PAN en dehors du fait que la corticothérapie est poursuivie à faible dose (5 à 10 mg/j) en raison de la persistance habituelle de l’asthme après la guérison de la vascularite.